
Forschung und Innovation als Standortfaktor – Rechtliche Weichenstellungen für Life Sciences made in Europe
Forschung und Innovation gelten als Schlüsselressourcen europäischer Wettbewerbsfähigkeit – und als wesentliche Standortfaktoren für forschungsintensive Branchen wie die Life Sciences. Die Europäische Union beantwortet diesen Befund zunehmend mit einem vielschichtigen Regelungs- und Maßnahmenkatalog: Neben horizontalen und sektoralen Rechtsrahmen treten datenrechtliche Infrastrukturen sowie operative Initiativen, die Verfahren beschleunigen und die Translation von Forschung in Versorgung und Markt erleichtern sollen.
Der Beitrag zeichnet diese Entwicklung entlang dreier eng miteinander verflochtener Ebenen nach, die sich gerade im Life-Sciences-Sektor nicht getrennt bewerten lassen, sondern erst im Zusammenspiel ihre Standortwirkung entfalten:
- Horizontale, sektorübergreifenden Weichenstellungen, mit denen die EU regulatorische Kohärenz, Skalierbarkeit und Investitionsfähigkeit im Binnenmarkt stärken will. Diese Initiativen setzen branchenübergreifend an, etwa durch den Abbau von Fragmentierung, mehr Rechtsklarheit und innovationsfreundlichere Verwaltungs- und Genehmigungsprozesse.
- Sektorspezifische Reformen des Biotech-, Pharma- und Medizinprodukterechts. Diese Ebene greift unmittelbar in die Wertschöpfungskette von Forschung über klinische Entwicklung bis hin zu Zulassung und Marktzugang ein.
- Entwicklung der datenrechtlichen und technischen Infrastruktur für den Zugang zu Gesundheitsdaten, die als Ressource für moderne Forschung, Real-World-Evidence und KI-gestützte Entwicklungsprozesse von erheblicher Bedeutung sind.
Übergreifend steht der Leitgedanke im Mittelpunkt, dass Regulierung nicht allein Schutzstandards sichern, sondern zugleich als „Enabler“ eines wettbewerbsfähigen Innovationsökosystems wirken soll – insbesondere durch beschleunigte Verfahren, eine bessere Abstimmung benachbarter Regime (etwa bei Arzneimittel/Medizinprodukte-Kombinationen) sowie eine praxistaugliche Ausgestaltung von Anreiz- und Schutzmechanismen.
I. Innovation als Regulierungsziel: Die neue horizontale Architektur der EU
Die nachfolgenden horizontalen Initiativen der EU bilden den übergreifenden Ordnungsrahmen der aktuellen Reformagenda, mit der Forschung, Innovation und industrielle Wettbewerbsfähigkeit systematisch gestärkt werden soll. Regulatorische Rahmenbedingungen, Dateninfrastrukturen und industriepolitische Instrumente sollen nicht länger isoliert betrachtet werden, sondern in einem integrierten Ansatz zusammenwirken müssen, um Europas Attraktivität als Forschungs- und Innovationsstandort zu sichern.
Politische Leitinitiativen, die programmatisch vorgeben, welche Ziele die nachfolgenden Rechtsakte absichern sollen, sind insbesondere die „Life-Sciences Strategie 2030“ und der „Kompass für eine wettbewerbsfähige EU“:
- Die Life-Sciences-Strategie 2030 dient als strategischer Referenzrahmen für horizontale Initiativen und entfaltet als Soft-Law-Instrument Vorwirkungen für spätere Legislativakte. Ziel ist, Europa bis 2030 zum weltweit attraktivsten Standort für Biowissenschaften zu machen. Sie verfolgt einen ganzheitlichen Ansatz entlang der gesamten Wertschöpfungskette – von der Grundlagenforschung über die klinische Entwicklung bis hin zu Marktzugang und Versorgung. Neben gezielten Investitions- und Förderinstrumenten adressiert die Strategie vor allem regulatorische Engpässe, lange Genehmigungszeiten und mangelnde Kohärenz zwischen benachbarten Rechtsregimen, etwa im Zusammenspiel von Arzneimittel-, Medizinprodukte- und Chemikalienrecht. Hinzu treten Initiativen zum besseren Zugang zu Gesundheitsdaten, zur Stärkung von Public-Private-Partnerships sowie zur Förderung von Translation und Skalierung innovativer Technologien. Die Life-Sciences-Strategie fungiert damit als sektorspezifischer Pilot für eine innovationsfreundlichere Binnenmarktarchitektur, die Schutzstandards wahrt, aber stärker auf Geschwindigkeit, Planbarkeit und Investitionssicherheit ausgerichtet ist.
- Flankiert wird dieser Ansatz durch den „Kompass für eine wettbewerbsfähige EU“, der die industrie- und innovationspolitischen Ambitionen der Union in eine breitere wirtschaftsstrategische Agenda einbettet. Der Kompass zielt darauf, strukturelle Standortnachteile Europas gegenüber den USA und China zu adressieren, insbesondere bei Kapitalmobilisierung, Skalierung von Start-ups und Deep-Tech-Unternehmen sowie bei der Komplexität regulatorischer Vorgaben. Er verbindet Binnenmarktintegration, Vereinfachung und gezielte Industriepolitik mit dem Anspruch, Innovation als Leitmotiv der europäischen Regulierung zu verankern. Dabei rückt weniger die Schaffung immer neuer Förderprogramme als vielmehr die Modernisierung bestehender Rechtsrahmen in den Vordergrund, um Fragmentierung abzubauen und innovationshemmende Hürden zu senken.
Zur Schließung der Innovationslücke kündigt der Kompass u. a. einen European Innovation Act, den ERA Act und ein „28th Regime“ an, deren konkrete Ausgestaltung und Rechtswirkung noch offen sind.
- Der European Innovation Act zielt auf einen horizontalen, sektorübergreifenden Rechtsrahmen zur strukturellen Stärkung von Innovations- und Wettbewerbsfähigkeit. Er soll bestehende Regulierung innovationskompatibel verzahnen, administrative Hürden abbauen und unionsweit vergleichbare Rahmenbedingungen schaffen; der Erlass als Verordnung ist für das erste Quartal 2026 vorgesehen.
- Der European Research Area Act adressiert die vorgelagerte Ebene der Forschungs- und Investitionsbedingungen. Er soll öffentliche und private F&E-Investitionen erhöhen, das 3-%-BIP-Ziel bekräftigen sowie die Abstimmung zwischen EU und Mitgliedstaaten und die grenzüberschreitende Forschung, Infrastruktur- und Technologietransfers stärken; die öffentliche Konsultation läuft bis Januar 2026.
- Das 28th Regime richtet sich auf die unternehmerische Umsetzung, Skalierung und Finanzierung innovativer Geschäftsmodelle und ist als optionaler, unionsweit einheitlicher Rechtsrahmen geplant, um regulatorische Fragmentierung abzubauen und grenzüberschreitende Gründung, Wachstum und Investitionen zu erleichtern; ein Legislativvorschlag wird für das erste Quartal 2026 erwartet.
II. Sektorale Weichenstellungen für Pharma, Biotech und die Medizinprodukte-Branche
Während die horizontalen Initiativen den institutionellen und rechtspolitischen Rahmen abstecken, greift die Europäische Kommission mit dem neuen Biotech Act, dem Pharma Package sowie den geplanten Änderungen der MDR und IVDR gezielt in jene Regulierungsbereiche ein, die für Forschung, klinische Entwicklung und Marktzugang in den Life Sciences maßgeblich sind. Gemeinsam ist diesen Reformen der Versuch, hohe Schutzstandards mit beschleunigten Verfahren, risikoproportionierter Regulierung und verbesserten Investitionsanreizen zu verbinden.
1. Pharma Package
Den Ausgangspunkt der sektoralen Reformagenda bildet die systemische Neuordnung des allgemeinen Arzneimittelrechts durch das neue Pharma Package. Der Rat und das Europäische Parlament haben sich am 11. Dezember 2025 vorläufig zum Pharma Package geeinigt. Dieses umfasst im Wesentlichen sowohl eine neue Verordnung als auch eine neue Richtlinie, durch die die bislang auf dem Gebiet des Arzneimittelrechts bestehenden Rechtsakte der EU zum Teil geändert und zum Teil ersetzt werden sollen. Damit handelt es sich um die größte Reform des Arzneimittelrechts seit über 20 Jahren. Die Reform verfolgt den Zweck, einen gerechten Zugang zu sicheren, wirksamen und erschwinglichen Arzneimitteln in der gesamten EU zu gewährleisten und zugleich die Wettbewerbsfähigkeit der pharmazeutischen Industrie zu stärken. Damit rückt sie jene Anreiz- und Schutzmechanismen in den Mittelpunkt, die für Investitionsentscheidungen, Standortwahl und Entwicklungsstrategien im gesamten Arzneimittelsektor zentral sind. Nach der erzielten Einigung sind die Kernelemente des Pharma Package die Folgenden:
- Die Fristen des Unterlagen- und Vermarktungsschutzes werden von der bisherigen „8+2+1-Regel“ auf eine „8+1+1+1-Regel“ umgestellt: Acht Jahre Unterlagenschutz bleiben bestehen, gefolgt von einem einjährigen Vermarktungsschutz, der um jeweils ein Jahr verlängert werden kann, wenn entweder ein „unmet medical need“ adressiert wird oder das Produkt der Behandlung einer Kombination von Erkrankungen dient und bestimmte Studien durchgeführt sowie Antragsfristen eingehalten wurden. Eine Verlängerung der Vermarktungsschutzfrist um ein Jahr ist zusätzlich möglich im Fall einer therapeutischen Zusatzindikation mit erheblichem klinischem Nutzen.
- Mitgliedstaaten können von Zulassungsinhabern ausreichende Lieferungen besonders wichtiger Arzneimittel verlangen.
- Die „Bolar-Ausnahmeregelung“ wird präzisiert und ihr Anwendungsbereich auf die Einreichung von Angeboten für öffentliche Aufträge ausgeweitet.
- Zur Bekämpfung antimikrobieller Resistenzen wird ein übertragbarer Gutschein eingeführt, der als Anreiz für die Entwicklung prioritärer Antibiotika dient und ein zusätzliches Jahr Marktschutz für ein Arzneimittel nach Wahl des pharmazeutischen Unternehmens (ausgenommen werden jedoch Arzneimittel, die in den vorangegangenen vier Jahren einen Jahresumsatz von mehr als 490 Mio. Euro brutto erzielt haben).
- Für Arzneimittel für seltene Leiden wird der Basisschutz der Marktexklusivität von 10 auf 9 Jahre verkürzt. Für die neu geschaffene Kategorie der bahnbrechenden Arzneimittel für seltene Leiden gilt eine verlängerte Marktexklusivität von 11 Jahren.
- Verkürzung des regulären Zulassungsverfahrens von 210 auf 180 Tage.
Die formelle Billigung durch Rat und Parlament steht noch aus. Ist sie erfolgt, werden sowohl die neue Verordnung als auch die neue Richtlinie im Amtsblatt der EU veröffentlicht werden und damit – unter Berücksichtigung etwaiger Übergangsvorschriften – in Kraft treten. Auch wenn eine abschließende Bewertung der Neuerungen erst mit Vorliegen der endgültigen Verordnungs- und Richtlinientexte möglich sein wird, erscheint die Einigung zwischen Rat und Kommission deutlich innovationsfreundlicher und als der in 2023 vorgelegte Vorschlag des Pharma Packages der Europäischen Kommission.
2. Biotech Act I
Der Biotech Act knüpft an die Reform des Arzneimittelrechts an und adressiert gezielt den innovationsintensiven Kernbereich der biopharmazeutischen Forschung und Entwicklung. Mit dem am 16. Dezember 2025 vorgelegten Verordnungsentwurf setzt die Kommission ein klares Signal zur Stärkung des europäischen Biotechnologie- und Biomanufacturing-Standorts und zielt auf innovationsfreundliche, wissenschaftsbasierte und planbare regulatorische sowie finanzielle Rahmenbedingungen. Der Vorschlag ist Teil der Life-Sciences-Strategie 2030, befindet sich im ordentlichen Gesetzgebungsverfahren und dürfte frühestens Ende 2026 verabschiedet werden. Bereits politisch avisiert ist zudem ein weiterer Regelungskomplex („Biotech Act II“), der über den primär gesundheitsbezogenen Fokus des ersten Teils hinausgehen und zusätzliche Bereiche der Biotechnologie adressieren soll.
Inhaltlich erfasst der Biotech Act den gesamten Lebenszyklus gesundheitsbezogener Biotechnologie – von Forschung und Finanzierung über klinische Entwicklung bis zu Herstellung und Marktzugang. Vorgesehen sind beschleunigte und vereinfachte Genehmigungsverfahren, der Ausbau industrieller Produktionskapazitäten, die Reduzierung kritischer Lieferkettenabhängigkeiten sowie gezielte Finanzierungsinstrumente für späte Entwicklungsphasen und den industriellen Scale-up. Zentrale Elemente sind gesetzlich definierte Kategorien „strategischer“ und „hochwirksamer“ Biotech-Projekte mit prioritären Verfahren, administrativer Unterstützung und verbessertem Zugang zu Fördermitteln. Flankierend setzt der Entwurf Akzente bei Digitalisierung, KI-Einsatz, regulatorischen Unterstützungsstrukturen (Netzwerke, Anlaufstellen, Sandboxes) sowie punktuellen Anpassungen von Schutz- und Anreizmechanismen, insbesondere beim SPC. Der Ansatz ist ausdrücklich sektorübergreifend und bezieht neben Human- auch Tierarzneimittel ein.
Besonders weitreichend sind die geplanten Änderungen für klinische Prüfungen nach der CTR:
Die Genehmigungsdauer multinationaler Studien soll von 106 auf 75 Tage verkürzt werden. Für ATMP-Studien entfällt die Verlängerungsoption um 50 Tage. Auch bei wesentlichen Änderungen werden die Prüfzeiten deutlich reduziert. Der reporting Member State wird als federführender Taktgeber gestärkt, Einwände anderer Mitgliedstaaten enger gefasst und EU-harmonisierte Templates eingeführt. Ergänzend sollen dezentralisierte Studien erleichtert, als neue Kategorie „minimal-interventionelle klinische Prüfungen“ definiert werden, kombinierte Arzneimittel-/Medizinprodukte bzw. IVD-Studien vereinfacht, Digitalisierung und KI regulatorisch integriert und die datenschutzrechtliche Grundlage für die Datenverarbeitung einheitlich geregelt werden. Insgesamt wird die CTR nicht neu konzipiert, aber gezielt beschleunigt, harmonisiert und risikoproportionierter ausgestaltet – mit klaren Standortimpulsen für multinationale und ATMP-Studien.
3. MDR / IVDR 2.0
Während Pharma Package und Biotech Act primär das Arzneimittel- und Biopharmarecht neu ordnen, erfasst die Reform der Verordnungen (EU) 2017/745 über Medizinprodukte („MDR“) und 2017/746 über In-vitro Diagnostika („IVDR“) den angrenzenden, zunehmend innovationsgetriebenen Bereich der Medizinprodukte und In-vitro-Diagnostika. Nach einer Targeted Evaluation (Dezember 2024 bis März 2025) und einem Call for Evidence (September bis Oktober 2025) legte die Europäische Kommission am 16. Dezember 2025 ihren Änderungsvorschlag zur MDR und IVDR; die derzeit laufende Konsultationsphase dauert bis 16. März 2026. Ziel ist es, den Rechtsrahmen für Medizinprodukte und IVD zu straffen, zukunftssicher und kosteneffizienter zu gestalten, Verwaltungsaufwand zu reduzieren und Zertifizierungsverfahren vorhersehbarer zu machen, ohne das hohe Niveau von Gesundheits- und Patientenschutz zu senken. Zugleich soll die Harmonisierung vertieft werden, um Wettbewerbsfähigkeit und Innovation im EU-Markt zu stärken.
Die wesentlichen Änderungen lassen sich wie folgt skizzieren:
- Vereinfachung und Verhältnismäßigkeit
- Person responsible for regulatory compliance (PRRC): Aufhebung der detaillierten Qualifikationsanforderungen für die PRRC und Aufhebung der Verpflichtung, dass KMU, die auf eine externe PRRC angewiesen sind, diese „ständig und kontinuierlich” zur Verfügung haben müssen.
- Gültigkeit von Zertifikaten und Rezertifizierung: Die maximale Gültigkeitsdauer von Zertifikaten (derzeit 5 Jahre) wird abgeschafft. Anstelle einer erneuten Zertifizierung der Produkte führen Benannte Stellen während der Gültigkeitsdauer des Zertifikats regelmäßige Überprüfungen durch, die dem Risiko des Produkts angemessen sind.
- Etablierte Technologien: Für Produkte, die weniger strengen Anforderungen unterliegen, wird eine Definition des Begriffs „bewährtes technisches Produkt“ eingeführt.
- Klassifizierungsregeln nach Annex VIII der MDR: Einige Klassifizierungsregeln werden angepasst, was zu niedrigeren Risikoklassen für bestimmte Produkte führt, wie beispielsweise wiederverwendbare chirurgische Instrumente, Zubehör für aktive implantierbare Produkte und Software.
- Verringerung des Verwaltungsaufwands
- Zusammenfassung der Sicherheit und (klinischen) Leistung: Der Umfang der Produkte, für die der Hersteller eine Zusammenfassung der Sicherheit und (klinischen) Leistung (SS(C)P) vorlegen muss, wird auf diejenigen Produkte beschränkt, für die die Benannte Stelle eine Bewertung der technischen Dokumentation durchführen muss.
- Regelmäßiger Sicherheitsbericht: Die Häufigkeit, mit der Hersteller verpflichtet sind, regelmäßige Sicherheitsberichte (PSUR) zu aktualisieren, wird reduziert. Die Überprüfung der PSUR durch die benannte Stelle wird Teil ihrer Überwachungsaktivitäten sein.
- Änderungen nach der Zertifizierung: Die Benannte Stelle muss zwischen Änderungen am Qualitätsmanagementsystem oder am zugelassenen Produkt unterscheiden, die Hersteller ohne vorherige Meldung, ohne vorherige Genehmigung oder nur nach Genehmigung durch die Benannte Stelle umsetzen können.
- Innovation und Verfügbarkeit von Produkten für besondere Patientengruppen
- In-house devices: Die Bedingungen für die Herstellung und Verwendung innerhalb von Gesundheitseinrichtungen werden flexibler gestaltet. Gemäß der IVDR entfällt die Bedingung, dass es kein gleichwertiges Produkt auf dem Markt gibt.
- Konformitätsbewertungsverfahren für bahnbrechende Medizinprodukte oder solche für seltene Leiden: Nach der Einstufung durch ein Expertengremium werden bahnbrechende Medizinprodukte und Orphan-Medizinprodukte einer vorrangigen und fortlaufenden Prüfung unterzogen.
- Einrichtung Regulatorischer Sandboxes, um den Anforderungen neuer Technologien gerecht zu werden
- Aufbereitung von Einmal-Produkten: Die Hersteller sind verpflichtet, eine Begründung für die Angabe „Einmalgebrauch“ anzugeben. Alle Produkte, die nicht für den Einmalgebrauch bestimmt sind, können gemäß den Anweisungen des Herstellers wiederaufbereitet werden. Eine Person, die ein Einmalprodukt vollständig wiederaufbereitet, gilt als Hersteller des vollständig wiederaufbereiteten Produkts.
- Vorhersehbarkeit und Kosteneffizienz der Zertifizierung
- Konformitätsbewertungsverfahren: Die Beteiligung benannter Stellen an der Konformitätsbewertung von Produkten mit geringem und mittlerem Risiko (Klasse IIa und IIb sowie Klasse B und C) wird reduziert (technische Dokumentationsprüfung eines repräsentativen Produkts für eine generische Produktgruppe, für eine Kategorie oder für das gesamte Portfolio).
- Gebühren der Benannten Stellen sollen für KMU sowie Hersteller von Orphan-Produkten reduziert werden.
Insgesamt ist der Vorschlag zur Änderung der MDR und IVDR als ein wichtiger Schritt zu einem industriefreundlichen und innovationsfördernden Regulierungsrahmen zu sehen, wenn auch konkrete Entlastungen erst mittelfristig wirksam werden. Bis zu einer Verabschiedung der Änderungsverordnung, die laut Europäischer Kommission in Q2/2027 zu erwarten sein soll, ist jedoch weiterhin mit Anpassungen des Verordnungsvorschlags im Rahmen des laufenden Gesetzgebungsverfahrens zu rechnen, sodass Inhalt und Reichweite der angekündigten Regelungen sicherlich noch Änderungen unterliegen werden.
III. Rechtsrahmen für den Zugang zu Gesundheitsdaten zu Forschungszwecken
Neben der Reform der sektoralen Regime wird die Standortattraktivität zunehmend durch einen weiteren Engpass geprägt: die Verfügbarkeit, Interoperabilität und rechtssichere Nutzbarkeit von Gesundheitsdaten.
Mit dem European Health Data Space (EHDS) etabliert die EU hierfür einen unionsweit harmonisierten rechtlichen, technischen und organisatorischen Rahmen für den sicheren und standardisierten Zugang zu elektronischen Gesundheitsdaten, sowohl für die sekundäre Nutzung (u. a. Forschung und Innovation) als auch für den verbesserten Zugriff der Patientinnen und Patienten auf ihre eigenen Daten (primäre Nutzung). Ziel ist der Abbau rechtlicher Fragmentierung, begrenzter Interoperabilität und uneinheitlicher Zugangsvoraussetzungen.
Die EHDS-Verordnung ist am 26. März 2025 in Kraft getreten und wird schrittweise umgesetzt; die meisten Verpflichtungen gelten erst vier Jahre später. Der erste Meilenstein der Umsetzung ist März 2027 mit dem Erlass wesentlicher Durchführungsakte.
In Deutschland wird der EHDS flankiert durch das Gesundheitsdatennutzungsgesetz (GDNG) vom 22. März 2024, welches die Nutzung von Gesundheitsdaten zu gemeinwohlorientierten Forschungszwecken und zur datenbasierten Weiterentwicklung des Gesundheitswesens als lernendes System regelt. Das GDNG sieht u. a. die Einrichtung einer zentralen Datenzugangs- und Koordinierungsstelle sowie einer dezentralen Gesundheitsdateninfrastruktur vor, um Zugangsbarrieren abzubauen.
Zudem wurde in Deutschland am 9. Oktober 2025 das Forschungsdatenzentrum Gesundheit (FDZ Gesundheit) eröffnet, welches am Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) angesiedelt ist. Das FDZ Gesundheit ermöglicht die Nutzung pseudonymisierter Gesundheitsdaten, insbesondere gesetzlicher Krankenversichertendaten, für ausgewählte sekundäre Verwendungszwecke in der Forschung. Diese nationale Einrichtung ist Teil der Umsetzungsstrategie zur Verwirklichung der EHDS-Ziele und soll den Zugang zu Gesundheitsdaten auch technisch erleichtern.
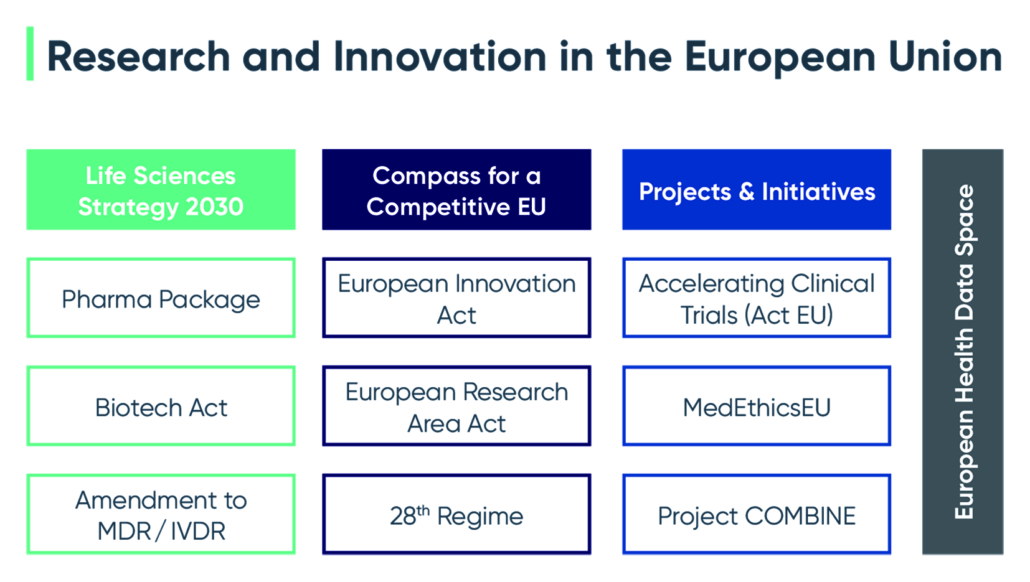
IV. Koordination statt Kodifikation: Projekte und Initiativen zur Förderung von Forschung und Innovation
Neben den legislatorischen Bestrebungen werden von der Europäischen Kommission, teils in Zusammenarbeit mit den Mitgliedstaaten, auch eine Vielzahl von Projekten und Initiativen zur Förderung von Forschung und Innovation sowohl angestoßen als auch fortgeführt:
- Die Initiative „Beschleunigung klinischer Prüfungen in der Europäischen Union“ (ACT EU) ist eine gemeinsame Initiative der Kommission, der EMA sowie der Leiter der Heads of Medicines Agencies (HMA), die im Januar 2022 ins Leben gerufen wurde. Erklärtes Ziel ist es, die Entwicklung von hochwertigen, sicheren und wirksamen Arzneimitteln weiter zu fördern sowie die klinische Forschung besser in das europäische Gesundheitssystem zu integrieren. Hierfür soll die Art und Weise der Initiierung, Konzipierung sowie der Durchführung von klinischen Studien transformiert werden.
- Die Initiative MedEthicsEU wurde im Februar 2024 als eine Gruppe von nationalen Vertretern von Ethikkommissionen für die medizinische Forschung gegründet. Sie soll einen Beitrag zur Koordination der Arbeitsweisen der nationalen Ethikkommissionen und zur Angleichung der operativen Verfahren leisten. Hierfür werden gemeinsame Leitlinien, Mustervorlagen und Verfahren entwickelt sowie Anreize für deren Verwendung geschaffen, wodurch wiederrum die Transparenz und Vorhersehbarkeit der rechtlichen und ethischen Bewertung der klinischen Prüfungen gestärkt werden soll.
- Das COMBINE-Programm wurde im Juni 2023 von der Europäischen Kommission und den zuständigen Behörden der Mitgliedstaaten initiiert, um die Schnittstellen zwischen den getrennten Zulassungsregimen für Arzneimittel, Medizinprodukte und In-vitro-Diagnostika zu optimieren. In der EU sind die Zulassungsverfahren für Arzneimittel, Medizinprodukte und In-vitro-Diagnostika jeweils separat und mit speziellen Anforderungen geregelt. Im Hinblick auf die Zunahme von innovativen und personalisierten Behandlungen, im Rahmen derer die Anwendung von Arzneimitteln und Medizinprodukten kombiniert wird, verfolgt das COMBINE-Programm das Ziel, die Schnittstelle zwischen den verschiedenen Rechtsrahmen zu optimieren. Ziel ist es, regulatorische Herausforderungen bei kombinierten Studien zu identifizieren und Lösungen zu entwickeln. Erprobt wird insbesondere ein koordiniertes „all-in-one“-Bewertungsverfahren, das Genehmigungen für Arzneimittel und Medizinprodukte in mehreren Mitgliedstaaten in einem Verfahren bündelt und so den Verwaltungsaufwand für Sponsoren reduziert.
Fazit: Auf dem Weg zu „Life Sciences Innovations Made in Europe“
Die skizzierten Initiativen markieren einen bemerkenswerten Paradigmenwechsel in der europäischen Innovationspolitik. Erstmals wird Innovation nicht nur als Ergebnis guter Wissenschaft, sondern als eigenständiges Regulierungsziel verstanden, das systematisch in die Architektur des Binnenmarkts eingebettet wird. Die horizontalen Projekte – European Innovation Act, ERA Act und 28th Regime – zielen auf die strukturelle Entfragmentierung des Rechtsraums und die Schaffung unionsweit skalierbarer Rahmenbedingungen. Sie adressieren damit eine der zentralen Schwächen des europäischen Innovationssystems: die mangelnde Rechts- und Investitionskohärenz über nationale Grenzen hinweg.
Auf sektoraler Ebene greifen Biotech Act, Pharma Package sowie die Reform der MDR und IVDR gezielt in jene Abschnitte der Wertschöpfungskette ein, in denen sich regulatorische Steuerung und standortpolitische Wirkung in besonderer Weise überlagern: bei der Dauer und Vorhersehbarkeit von Genehmigungsverfahren, bei der Komplexität und Kohärenz der einschlägigen Regime, bei der Ausgestaltung von Schutz- und Anreizmechanismen sowie beim Zugang zu klinischen und realweltlichen Daten. Insbesondere die vorgesehenen Beschleunigungen im Recht der klinischen Prüfungen, die stärker risikoproportionierte Architektur des Medizinprodukterechts und die Neujustierung der Anreizsysteme im Arzneimittelrecht sind geeignet, die strukturellen Standortnachteile Europas zumindest teilweise zu korrigieren.
Flankiert wird diese regulatorische Neuordnung durch den Aufbau unionsweiter und nationaler Dateninfrastrukturen im Rahmen des European Health Data Space. Erstmals entsteht damit ein rechtlich harmonisierter Rahmen, der den großskaligen, grenzüberschreitenden Zugang zu Gesundheitsdaten nicht nur ermöglicht, sondern als integralen Bestandteil des Innovationssystems institutionell absichert. Diese Form „regulatorischer Infrastruktur“ wirkt nicht allein verfahrensbeschleunigend, sondern eröffnet qualitativ neue Entwicklungspfade – von der systematischen Nutzung von Real-World-Evidence über KI-gestützte Entwicklungsprozesse bis hin zu datengetriebenen Ansätzen personalisierter Medizin.
Ob Europa tatsächlich zum „weltweit attraktivsten Standort für Life Sciences“ werden kann, wird weniger von der Vielzahl der angekündigten Rechtsakte abhängen als von ihrer kohärenten, investitionsstabilen und international wettbewerbsfähigen Ausgestaltung. Entscheidend ist, ob es gelingt, Beschleunigung und Flexibilisierung der Verfahren mit einem verlässlichen Schutz geistigen Eigentums, praxistauglichen Anreizsystemen und einer Begrenzung zusätzlicher administrativer Lasten zu verbinden. Gerade im globalen Wettbewerb um „first launches“, klinische Studien und industrielle Wertschöpfung drohen inkonsistente Anreizstrukturen, überzogene Melde- und Transparenzpflichten oder faktische Verkürzungen effektiver Schutzfristen die Standortattraktivität zu unterminieren, statt sie zu stärken.
Gelingt hingegen eine widerspruchsfreie Verzahnung der Reformen – von der Arzneimittel- und Biotech-Regulierung über die Medizinprodukte bis hin zur Dateninfrastruktur –, könnte sich ein genuin europäisches Innovationsmodell herausbilden: getragen von hohem Schutzniveau und zugleich geprägt von regulatorischer Geschwindigkeit, Planungssicherheit und datenbasierter Entwicklung. In diesem Sinne markieren die aktuellen Weichenstellungen mehr als eine regulatorische Feinjustierung. Sie sind ein Testfall dafür, ob Europa in der Lage ist, ein innovationsfreundliches, rechtssicheres und investitionsattraktives Ordnungsmodell zu etablieren – oder ob sich die beabsichtigte Standortoffensive im Dickicht konkurrierender Zielsetzungen und zunehmender Bürokratisierung verliert.

Autor:
Irina Rebin
Salary Partnerin
München
Taylor Wessing Partnerschaftsgesellschaft mbB
 Uniklinikum Leipzig
Uniklinikum Leipzig Boehringer Ingelheim
Boehringer Ingelheim Proxygen GmbH
Proxygen GmbH